FORMATION ET DEVELOPPEMENT DE L’IMAGE PHOTOGRAPHIQUE
Auteur Pierre Glafkidès
Physique et chimie photographique

- Lois fondamentales
- Dissociation photochimique
- Réactions photochimiques organiques
- Réactions photochimiques inorganiques
- Phototropie
- Fluorescence et phosphorescence
La photochimie est l’étude des transformations chimiques provoquées par la lumière. Le phénomène photochimique comporte deux phases principales : 1° réception de l’énergie lumineuse ; 2° réaction chimique proprement dite. Suivant que l’on a affaire à une substance unique ou à un système de plusieurs corps en présence, on assiste soit à une décomposition de la substance en ses éléments (photolyse), soit à une combinaison de plusieurs corps en un seul (photosynthèse).
Lois fondamentales
1- Loi d’absorption de Grotthus-Draper :
Une radiation ne peut provoquer une action chimique que si elle est absorbée par un corps (ou un système de corps). Sinon, il ne peut y avoir transmission d’énergie lumineuse.
Il convient de remarquer que les radiations qui constituent la couleur d’un corps sont justement celles qui ne sont pas absorbées : elles sont donc sans effet sur celui-ci. Par contre les radiations complémentaires de cette couleur sont absorbées et sont susceptibles d’action. Par exemple, une substance de couleur verte renvoie le vert mais absorbe le bleu et le rouge : elle ne pourrait être décomposée que par ces deux dernières couleurs.
Toutefois une radiation absorbée n’agit pas nécessairement sur la substance qui la reçoit. 11 faut de plus, pour cela, qu’elle satisfasse à d’autres conditions.
2- Loi énergétique
Pour agir efficacement, une radiation lumineuse doit posséder une énergie au moins égale à celle nécessitée par la transformation chimique. On sait que les radiations sont d’autant plus énergiques que leurs longueurs d’ondes sont plus courtes (ou que leurs fréquences v sont plus élevées), l’énergie transportée par un photon étant donnée par l’expression :

Ou h est la constante de Planck, égale à 6,62 10-27 erg/s
A une molécule-gramme contenant N molécules correspond un photon-gramme contenant N photons (*) soit 603.1021 photons dont l’énergie totale est : U = Nhc/λ. ergs. En faisant la transformation en calories équivalentes on obtient l’énergie calorifique d’un photon-gramme par la formule :
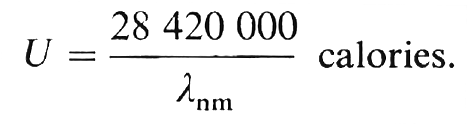
Le tableau suivant donne une idée de l’énergie des différentes radiations :

3- Loi d’équivalence photochimique (ou loi d’Einstein)
A chaque photon absorbé correspond une molécule décomposée ou combinée.
Il est sous-entendu que le photon actif satisfait à la loi énergétique précédente.
Or, on constate que, pratiquement, le nombre de photons actifs absorbés dans une réaction chimique correspond rarement au nombre de molécules formées ou disparues. (*) N est le nombre d’Avogadro.
Si l’on appelle rendement quantique p le rapport du nombre de molécules décomposées au nombre de photons absorbés :
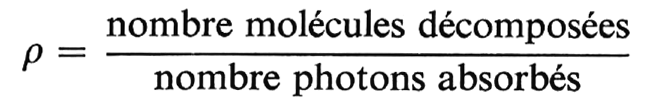
on obtient un rendement quantique qui varie dans de grandes limites, de 0,1 à 1 000 (et plus). Seules quelques réactions ont un rendement théorique égal à l’unité.
Pourtant cette contradiction ne met pas en cause la validité de la loi d’Einstein, et la raison des variations expérimentales est simple :
a) Quand la réaction chimique exige un apport extérieur d’énergie (réaction endothermique), et c’est le cas des halogénures d’argent, p est égal tout au plus à 1 ; en général il est plus faible, comme dans la décomposition photochimique du gaz chlorhydrique (1) car cette réaction est réversible

Pour décomposer l’ammoniac NH3 en azote et hydrogène, par les rayons ultraviolets, il faut quatre photons par molécule (ρ = 0,25).
Suivant la longueur d’onde on peut faire varier l’équilibre photochimique dans un sens ou dans l’autre. Telle est la réaction réversible (2) :
acide maléique <=> acide fumarique
où avec l’ultraviolet λ = 313 nm il y a 44 % d’acide maléique et 56 % d’acide fumarique tandis qu’avec une onde plus courte λ = 200 nm l’acide maléique se reforme avec 75 % d’acide maléique et 25 % d’acide fumarique. Dans le premier cas, le rendement quantique est de 0,03 (30 photons) tandis qu’il monte à 0,1 (10 photons) pour la réaction inverse (reformation de l’acide maléique).
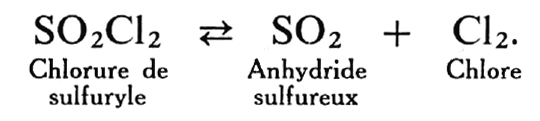
On observe un phénomène analogue dans la formation du chlorure de sulfuryle SO2C12 à partir de l’anhydride sulfureux et du chlore :
b) Quand les radiations absorbées provoquent d’abord une activation de la molécule, celle-ci réagit ensuite sur une seconde molécule neutre pour donner des produits de décomposition, suivant le schéma ci-dessous (3) :

Le rendement quantique est alors voisin de 2. Exemple la décomposition par la lumière ultraviolette (260 à 360 nm) du peroxyde d’azote NO2 en oxyde azotique NO et oxygène

pour λ = 400 nm le rendement p tombe à 0,75 et devient nul dans le spectre visible.
c) Quand la réaction est franchement exothermique (avec dégagement de chaleur) le rendement quantique devient très élevé. Dans ce cas, il suffit que la lumière fasse démarrer la réaction pour que celle-ci puisse se continuer d’elle-même à l’aide de sa propre chaleur dégagée, suivant une chaîne.
La combinaison du chlore avec l’hydrogène, en présence d’une trace de vapeur d’eau, par la lumière, est un exemple classique : ρ peut atteindre 100 000 (il varie avec la pression et le volume du récipient). On peut citer toute une série de combinaisons analogues :

ainsi que l’union du chlore avec de nombreuses substances organiques : alcool, éther, acide acétique, benzène, toluène, etc. Avec ce dernier ρ = 30.
Le phénomène photochimique exige quelquefois, pour avoir lieu, la présence d’un sensibilisateur : exemples, l’action de l’hydrogène sur le benzène (4) et les réactions photochimiques du propane (5) catalysées par le mercure. La photo-décomposition de l’acide oxalique est sensibilisée par les ions uranyle : il y a là un transfert d’électrons des ions uranyle excités, aux ions oxalate. D’autres réactions sont sensibilisées par l’oxyde de zinc. L’intensité lumineuse est quelquefois importante à considérer : ainsi des décharges électriques de 10 kW, ayant une durée de 0,1 à 3 ms produisent des substances différentes de celles obtenues avec des rayonnements d’intensité moyenne (6).
Dissociation photochimique.
La décomposition photochimique d’une molécule est révélée par son spectre d’absorption.
Un spectre comporte alors deux parties qui se touchent : un spectre discontinu formé de raies fines d’absorption qui correspondent à une vibration des noyaux des atomes dans la molécule. Un spectre continu dont l’absorption varie d’une manière continue.
Les mouvements vibratoires sont tels que les atomes oscillent autour de leur position d’équilibre et s’échappent dès que l’énergie vibratoire dépasse une certaine amplitude : la molécule se trouve alors dissociée.
Donc le spectre d’une substance présente, d’un côté, des bandes fines d’absorption qui, d’abord nettement bien séparées, finissent par se toucher vers les courtes longueurs d’onde, jusqu’à se fondre en un spectre continu. La région de séparation des deux formes de spectre porte le nom de limite de convergence. Or, seules les radiations faisant partie du spectre continu ont le pouvoir de décomposer la substance. Et si l’on élève la température, les bandes les plus proches du spectre continu se fondent avec celui-ci, et les radiations appartenant à ces bandes, auparavant sans action, deviennent actives.
Ce phénomène, découvert par Victor Henri, s’appelle « prédissociation ». Les exemples en sont nombreux : photolyse de l’aldéhyde acétique (λ efficace < 315 nm), de l’aldéhyde benzoïque (λ efficace < 250 nm), du sulfure de carbone CS2 (deux limites de convergence dans l’ultraviolet), de l’anhydride sulfureux SO2 (également deux limites correspondant à deux régions d’absorption), du peroxyde d’azote NO2 (λ < 400 nm et λ < 245), etc.
Les bandes fines d’absorption correspondent à une vibration d’atomes ou de groupes d’atomes, qui s’annulent à la limite de convergence, pour les radiations actives. Les molécules se trouvent alors en état de predissociation ou état d’excitation électronique, sans dissociation réelle, par absorption de photons.
Réactions photochimiques organiques
Tous les types connus de réactions organiques peuvent être sujets à l’influence de la lumière. Nous n’en citerons que quelques-uns.
Décompositions
Acétone (7), acétamide (8), formiate de méthyle (9), mercaptans (10) : à 253,7 nm un quantum décompose 1,7 molécule de mercaptan. La roténone (11) est également susceptible de décomposition photochimique. Il en est de même des dérivés nitrés du benzène selenié : nitrosélénocyanure, nitrodiséléniure, acide nitrosélénieux, acide nitrosélénique, naphtalène-1,4 (ou 1,8) nitroselenieux ou 1,8 nitrosélénique (11 bis). Les dérivés du sélénium sont plus sensibles que les dérivés correspondant du soufre.
Hydrolyses
Tétrachlorure de carbone en acide chlorhydrique et acide carbonique (12), acides mono et dichloracétiques en acides glycolique et glyoxylique (13), cyclohexanone en acide caproïque (14).
Oxydations.
Sous l’influence de la lumière, en présence d’oxygène, les alcools, les
phénols, les cétones, les aldehydes, les acides s’oxydent. Certains di- et triarylméthanes sont transformés en peroxydes : le phénomène est particulièrement intense pour le 9-phénylxanthène (15). De nombreuses photo-oxydations sensibilisées ont été appliquées à la préparation de composés organiques (16).
Réactions d’addition
avec les halogènes, l’ammoniac, l’aniline : dans ce dernier cas on obtient des anilides.
Nitrosations.
Le chlorure de nitrosyle NOCI est un agent de nitrosation puissant lorsque la réaction s’effectue en présence d’une lumière intense. C’est ainsi que le cyclohexane mis en présence de chlorure de nitrosyle sous le faisceau d’une lampe à mercure de 10 kW se transforme en cyclohexanone-oxime
C6H12+NOC1→C6H10= NOH+HC1.
Celle-ci sert ensuite à fabriquer du caprolactame CO(CH2)5NH matière première du Nylon-6 (16 bis).
Isomérisations et polymérisations.
L’o-nitrobenzaldéhyde, en solution dans le benzène ou l’éther, se transforme en acide o-nitrosobenzoïque. L’o-nitrobenzylidène-acétophénone forme de l’indigo (17) :

L’acétylène se polymérise en benzène (18) (il en est de même pour ses dérivés). Le stilbène se dimérise en distilbène. L’anthracène, le phénylacétaldéhyde, l’acide cinnamylidènemalonique, l’∝-naphtoquinone, etc., donnent aussi un dimère.
L’acrylate et le méthacrylate de méthyle se polymérisent en présence de benzoïne ou de diacétyle agissant comme catalyseurs. On peut obtenir ainsi des reliefs pour l’impression (19). Sous l’influence des rayons ultraviolets, de nombreux polymères subissent, par contre, une dépolymérisation marquée. Ainsi on constate une diminution de viscosité des solutions de nitrate et d’acétate de cellulose (20), ou de méthyl-cellulose (21). Le rayonnement de ). inférieure à 340 nm dégrade la plupart des fibres. Entre 340 et 400 nm la réaction doit être souvent activée par l’humidité et l’oxygène atmosphériques ou des traces de métaux.
Réactions photochimiques inorganiques.
Les modifications allotropiques de certains corps simples, par la lumière, sont des opérations banales en chimie minérale : le phosphore blanc se transforme en phosphore rouge, l’arsenic jaune en arsenic gris, l’antimoine jaune en antimoine noir, le selenium rouge en selenium noir. Le produit final est toujours le plus stable. Les dérivés hydrogénés et les dérivés sulfurés de tous ces corps sont de même sensibles à la lumière. La photolyse de l’acide azothydrique et des azotures alcalins a été aussi étudiée (22) : c’est une réaction par chaîne, le corps intermédiaire étant de l’azote activé.
La plupart des sels métalliques sont photosensibles. Les sulfures en suspension dans l’eau peuvent subir une oxydation lente. Une coloration bleue a été obtenue avec le molybdate d’ammonium ou le tungstate de sodium en milieu chlorhydrique.
Ce qui distingue le comportement des substances salines de celui des composés précédemment signalés, c’est essentiellement leur caractère ionique. Alors que dans les systèmes gazeux seules des molécules entrent en jeu, ici nous n’avons à considérer que le mouvement des électrons de valence, car les sels métalliques, à l’état cristallisé ou en solution aqueuse, se présentent à l’état d’ions.
Parmi ces décompositions photochimiques, les plus remarquables sont celles des dérivés des éléments suivants : fer, chrome, manganèse, cobalt, urane, cuivre, mercure et surtout argent. La réaction est souvent réversible :

Si on absorbe le chlore au fur et à mesure de sa formation, la réaction n’a lieu que dans le sens de la photolyse (décomposition).
La sensibilité des sels de fer à la lumière a donné lieu à quelques applications que nous étudierons plus tard ; de même pour les sels de chrome.
Comme les sels ferriques, les sels manganiques Mn+++ et cobaltiques Co+++ sont ramenés par la lumière à l’état de sels manganeux Mn++ et cobalteux Co++, par perte, chacun, d’un électron de valence.
Le chlorure cuivreux CuCI se colore en violet-brun, tandis que le chlorure cuivrique CuC12 en solution alcoolique est réduit en chlorure cuivreux. De même, les sels d’urane, de plomb et de thallium sont sensibles à l’action de la lumière.
Parmi les sels de mercure, le nitrate mercureux HgNO3, le tartrate mercureux basique, l’oxalate mercureux basique et notamment l’oxalate de mercure-ammonium, présentent des transformations intéressantes.
Réaction d’Eder
Un mélange de chlorure mercurique HgC12 et d’oxalate d’ammonium en solution aqueuse se décompose très rapidement en chlorure mercureux insoluble HgC1 (calomel) qui précipite ; cette réaction très sensible peut servir de photomètre. Le mécanisme serait le suivant (23) :

Les halogènes accélèrent considérablement la réaction (23 bis). D’autre part, les cyanines à molécule plane et l’érythrosine la sensibilisent. Cette sensibilisation semble s’opérer par transfert d’énergie (24).
Phototropie
Certaines substances organiques, soumises à l’influence de la lumière, changent de couleur. Replacées dans l’obscurité, elles reprennent leur teinte première. C’est une variation réversible de couleur que l’on appelle phototropie ou photochromie.
Fluorescence et phosphorescence
Une substance fluorescente, soumise à l’action des rayons lumineux d’une certaine longueur d’onde (ultraviolet, par exemple), transforme ces rayons incidents en d’autres radiations émises de plus grande longueur d’onde, visibles.
C’est que la molécule M, ayant absorbé un quantum hv, passe par un état activé instable M’, puis se désactive en repassant à son état stable, avec émission d’un quantum hv’, de plus faible fréquence. Cet abaissement de fréquence s’explique par le fait que les molécules activées sont en continuelle rotation : la désactivation arrête le mouvement de la molécule puis produit l’expulsion d’un photon d’énergie inférieure à celle du photon absorbé.
La durée qui s’écoule entre l’activation et la désactivation varie de un cent millionième à un millième de seconde (sels d’uranyle) suivant la nature de la substance. Le rendement varie de même : le rapport photons émis/photons absorbés s’approche de l’unité pour la fluorescéine diluée. La lumière émise par un corps fluorescent peut être assimilée, suivant J. Perrin, à une « somme discontinue d’éclairs minuscules identiques ».
Dans le phénomène de la phosphorescence, la substance irradiée conserve sa luminosité pendant une durée de temps relativement longue (souvent plusieurs heures). Cette propriété est due à la présence de substances dites phosphorogènes, notamment des sulfures. La présence d’impuretés est indispensable pour entretenir l’instabilité des édifices cristallins ainsi déformés : cuivre, manganèse, chaux, terres rares.
Une molécule, activée par un photon absorbé, produit en se désactivant sur un niveau légèrement inférieur, une molécule instable. Celle-ci reçoit grâce à l’agitation thermique, un peu d’énergie, ce qui lui permet de redonner la forme activée en revenant sur son premier niveau d’activation. Le même cycle se répète jusqu’à ce que l’écart entre le niveau d’activation et le niveau de chute devienne trop grand pour être remonté.
Retrouvez moi sur Instagram
(1) Emschwiller : L’action chimique de la lumière : la photolyse (Centre de documentation chimique, Paris).
(2) Victor Henri : Les progrès récents de la photochimie, p. 3 (Centre de documentation chimique, Paris).
(3) Victor Henri, /oc. cit., p. 2.
(4) G.-S. Forbes et J.-E. Clive : JI. Amer. Chem. Soc., juin 1941, p. 1713-1716.
(5) B. de B. Darwent et E.-W.-R. Steacie : JI. Chem. Physics, décembre 1942, pp. 563-571.
(6) R.-G.-W. Norrish : Zeits. Elektrochem, octobre 1952, pp. 705-712.
(7) JI. Amer. Chem. Soc., juin 1944, pp. 974-977 et 1940, pp. 2052-2069.
(8) P. Amer. Chem. Soc., juillet 1941, pp. 2000-2002.
(9) JI. Amer. Chem. Soc., juin 1941, pp. 1521-1525.
(10) Trans. Faraday. Soc., février 1942, pp. 81-82.
(I 1) JI. Amer. Chem. Soc., juin 1941, pp. 1717-1718.
(I l bis) Koslov V.V. : Acta Chim. Hungar., 1952, t. 12, pp. 189-194 (en russe).
(12) Liebig Ann., t. 392, 1911, p. 222.
(13) Zeits. f phys. Chem., 1913, p. 371.
(14) Ber, t. 46, p. 3077.
(15) A. Schônberg et A. Mustafa : JI. Chenz. Soc., octobre 1945, pp. 657-660.
(16) G.-O. Schenck : Angew. Chemie, janvier 1952, pp. 12-23.
(16 bis) Procédé P.N.C., de la Tokyo Rayon Co, à Tokyo (1962).
(17) Engler et Dorant : Ber, 1895, t. 28, p. 2497.
(18) B.-L. Dunicz : JI. Amer. Chem. Soc., septembre 1941, pp. 2461-2472.
(19) T.-S. Lawton et H.-K. Nason : Ind. and Eng. Chem., décembre 1944, pp. 1128-1130.
(20) Spitze, Mooradian, Hartigan et Hansen : JI. Amer. Chem. Soc., juin 1941, pp. 1576-1580.
(21) Gastes et Imp. Chem. Ind. : Br. Angl. 566 795 (1943).
(22) M. Bonnemay : Jl. Chim. Phys., avril 1944, pp. 56-81.
(23) G.-H. Cartledge : JI. Amer. Chem. Soc., 1941, pp. 906-912.
(23 bis) Marty et Gopale-Rao : Zeits. Physik. Chem., aout 1957, t. 207, pp. 167-179 (en anglais).
(24) Miss G. Kornfeld : Sc. et Ind. Phot., septembre 1952, p. 378.