FORMATION ET DEVELOPPEMENT DE L’IMAGE PHOTOGRAPHIQUE
Auteur Pierre Glafkidès
Physique et chimie photographique

- Contrôle des images lavées
- Elimination de l’hyposulfite par oxydation
- Analyse des bains de fixage
- Récupération de l’argent des bains d’hyposulfite
- Séchage
- Réticulation
- Glaçage et séchage des épreuves
- Examen analytique des dépôts formés sur les images
Lire la partie 1 de Fixage, lavage et séchage
Lire la partie 2 de Fixage, lavage et séchage
Contrôle des images lavées
Le fait que l’eau de lavage ne contient plus de trace d’hyposulfite ne signifie pas que l’image soit stable, mais seulement qu’il n’y a plus intérêt à prolonger le lavage. Il peut être donc nécessaire de vérifier s’il reste du fixateur dans la couche photographique lavée. Dans ce cas c’est l’échantillon même qui est traité par le réactif, par l’une des méthodes ci-après. Il faut toutefois faire des réserves : King et Henn avaient cru constater que les zones denses retiennent plus de thiosulfate que les zones claires. En réalité les différences sont dues aux valeurs de décomposition variables du thiosulfate dans les épreuves plus ou moins anciennes. Le thiosulfate se décompose en effet en trithionate puis tétrathionate sulfurants, d’où les erreurs (59).
a) Essai à l’iode (60) : Une solution O, 1 N d’iode est préparée avec 12,7 g d’iode et 30 g d’iodure de potassium dans Q.S. 1 000 ml d’eau, 25 ml de cette solution sont ensuite amenés à 1 000 ml. Puis on en dépose 5 gouttes aux quatre coins et aux dos d’une épreuve de 15 cm. Le lavage est insuffisant si la coloration des taches s’atténue rapidement. Cette décoloration correspond à 0,4 mg d’hyposulfite par décimètre carré.
b) Essai au chlorure mercurique (61) : Le réactif utilisé se compose d’une solution contenant 2,5 % de bromure de potassium et 2,5 % de chlorure mercurique (toxique). Un morceau de film de 25 x 25 mm, plié en deux, est introduit dans un tube à essai de 18 mm de diamètre. On y verse 10 ml de réactif, ainsi que dans sept autres tubes témoins contenant des quantités croissantes d’hyposulfite : 0, 0,005, 0,01, 0,02, 0,04, 0,1 et 0,2 mg, calculé anhydre (solution de 1/20 000 à 1/1 000). Après repos de 15 mn, on observe la turbidité due au précipité de chlorosulfure mercurique, et qui indique la présence d’hyposulfite.
c) Essai au nitrate d’argent applicable aux papiers (62) : On immerge pendant 3 mn l’échantillon dans une solution contenant 1 % de nitrate d’argent, 0,5 % d’acide sulfurique (en volume) ou 2 % d’acide acétique, puis 3 mn dans une solution à 5 % de chlorure de sodium, et finalement dans une solution fixatrice à 5 % d’hyposulfite et 2 % de sulfite ; après quoi on lave et sèche. La mesure de la densité de sulfure d’argent formé renseigne sur le degré d’élimination de l’hyposulfite.
Pour les films, la méthode a été modifiée de la manière suivante (63) : une goutte d’une solution contenant 0,75 % de nitrate d’argent et 3,5 % d’acide acétique, est déposée sur la face image du film. Après 2 mn on absorbe l’excès de liquide et compare la tache formée avec une échelle de quatre teintes imprimée sur une feuille d’acétate de cellulose. La tâche témoin la plus pâle correspond au traitement des films destinés aux archives. La méthode au nitrate d’argent connue sous le nom de CD-Index (Chemical-Densitometric Index) est celle indiquée par la norme ASA Z 38.8.25-1950. Elle est simple, à condition de disposer d’une aire de densité uniforme d’au moins 2 cm2. Elle est utilisable avec les émulsions pour la couleur.
Une autre méthode dite SID (Silver Ion Demand) consiste à traiter le phototype à 50° et pendant 30 mn par un excès de nitrate d’argent. Il se forme du sulfure d’argent. Le restant d’ions Ag+ est titré par de l’iodure de potassium à l’aide d’un potentiomètre.
Une technique, d’emploi général et plus complète, mais beaucoup plus complexe, a été adoptée par Pouradier et Chateau. Un échantillon mesuré de 12 cm2 et découpé en fragments est placé dans un flacon laveur. On y introduit deux fois 30 ml d’acide bromhydrique saturé d’hydroquinone, cela sous courant d’azote pur, H2S formé est entraîné ainsi pendant 2 h, dans deux flacons laveurs successifs contenant le premier 20 ml de nitrate d’argent M/2 000+5 ml d’ammoniaque 4 M+ 10 ml d’eau bidistillée et le second 35 ml d’eau bidistillée.
Les deux liquides sont transvasés dans un flacon à trois tubulures, l’une tenant une burette et une autre un tube plongeur pour l’azote. Au centre on place une électrode au sulfure d’argent et un pont rempli de nitrate de potassium saturé qui relie le liquide sulfhydrique à une électrode au calomel. Un potentiomètre sensible au millivolt mesure la force électromotrice. On ajoute donc au liquide de la thiourée en solution M/4 000 (préparée par dilution au moment de l’emploi) en suivant les variations de potentiel. Au point d’équivalence l’équilibre est très long à s’établir.
On détermine ainsi la quantité de sulfure formé durant la conservation. Pour le dosage du thiosulfate résiduel on recommence l’opération avec un échantillon découpé, 5 ml de solution de nitrate d’argent M/100. Le courant d’azote est envoyé pendant 10 mn, Le sulfure d’argent qui s’est ainsi formé est dosé alors comme précédemment.
Dans le choix de l’échantillon, tenir compte de la densité.
Pour obtenir l’électrode au sulfure d’argent, plonger une électrode d’argent, pendant 30 mn dans du sulfure d’ammonium 2 M. Quant à la purification de l’azote, elle se fait à l’aide de trois flacons laveurs contenant respectivement du nitrate d’argent M/100, du pyrogallol à 50 g dans la potasse 3 M et NaOH, N contenant 1 g d’hydroquinone par litre.
Le rôle de l’hydroquinone dans l’acide bromhydrique est de détruire les traces de brome.
d) Essai par les composés du sélénium : Henn et Crabtree ont par la suite préconisé l’emploi de composés du sélénium (64), déjà connus pour l’analyse des bains de fixage Ils ont l’avantage de ne pas réagir avec le barytage, et de produire des taches bien visibles. Pour préparer le réactif, faire une pâte avec 10 g de sélénium noir et 100 g de sulfite dans un peu d’eau à 90°. Ajouter 125 ml d’eau puis diluer au 1/20. La solution de sélénosulfate obtenue contient 0,2 % de Se. On peut la remplacer par un mélange d’hyposulfite et de sélénite (bain de virage au sélénium).
Elimination de l’hyposulfite par oxydation
Le meilleur procédé pour éliminer l’hyposulfite résiduel, dans une couche sensible, est le traitement à l’eau oxygénée, en milieu alcalin. Après lavage, immerger 6 mn à 18° dans le bain suivant ; préparé au moment de l’emploi :

Rinçage : 10 mn. Surface traitée : 50 dm2/I.
Avec 300 ml d’eau oxygénée à 10 volumes on peut traiter 80 dm2 (bain HE.2).
L’éliminateur à l’eau oxygénée peut produire soit un léger changement de ton de l’image, soit un léger jaunissement, soit le ramollissement de la gélatine, soit le collage du papier à la glaceuse. Pour éviter le changement de ton, on ajoute 1 g de bromure. Le jaunissement est détruit par deux minutes de rinçage dans l’acide acétique à 1 % ou le sulfite à 2 %, suivi de lavage. Même traitement pour raffermir la gélatine ramollie. Dans le cas de collage à la glaceuse, passer dans de l’alcool à 50 %.
L’hyposulfite peut être également détruit par oxydation à l’aide de persulfate de potassium ou d’ammonium à 1 %. en présence d’un tampon de citrate trisodique. Il se forme malheureusement des thionates qui sont susceptibles de sulfurer rapidement l’image argentique.
Analyse des bains de fixage
Argent : Pour doser l’argent contenu dans le bain fixateur on utilise la précipitation sous forme de sulfure : une prise d’essai de 50 ml et étendue à 100 ml est chauffée à 50°. On ajoute 30 ml de sulfure de sodium à 20 %. Après une demi-heure à 70-80°, le précipité se rassemble ; on filtre et le dissout dans 25 ml d’acide nitrique concentré bouillant. La solution diluée est précipitée par l’acide chlorhydrique : le chlorure d’argent formé est filtré, lavé, séché et pesé.
On peut aussi titrer l’argent de la solution nitrique par le sulfocyanure d’ammonium, en présence de sulfate ferrique.
Une autre méthode de dosage de l’argent, due à M. Abribat (65), consiste à traiter le fixateur usagé par du sélénosulfate ou du sélértotrithionate de sodium en présence de gélatine : il se forme une suspension colloïdale du sel d’argent correspondant, que l’on dose par colorimétrie dans un tube à essai calibré, la densité étant proportionnelle à la concentration en argent. 10 ml de réactif à 0,15 % contenant 0,4 % de gélatine sont additionnés de 0,5 ml de fixateur et de deux gouttes de lessive de soude, puis on compare avec un étalon.
On peut encore doser l’argent, dans les bains de fixage par le diéthylthiocarbonate de sodium, au moyen d’un potentiomètre, selon Kennula et Hulanicki (Chem. Anal. Warsaw, 1961, pp. 705-710, en anglais). On emploie une solution de réactif à 0,01 M, une électrode Ag-AgC1 et une électrode au calomel. On peut aussi opérer avec la thioacétamide selon la technique de Bush, Zuehlke et Ballard (voir analyse des émulsions), en présence d’éthylènediamine tétracétique (séquestrant) : on prélève une quantité de fixateur supposée contenir au moins 5 mg d’argent, ajoute 5 ml de solution de gélatine à 4 g/1, 25 ml de EDTA à 4 g/1 contenant 80 g de NaOH et titre avec de la thioacétamide à 0,01 N. Le potentiomètre a une électrode au sulfure d’argent obtenue par immersion d’une électrode d’argent dans du sulfure de sodium à 20 %, pendant 3 mn. Electrode de référence au calomel saturée.
Hyposulfite : Il faut d’abord transformer le sulfite présent, en un composé sans action sur l’iode. On traite, à cet effet, 2 ml de fixateur par 2 ml de formol dans 200 ml d’eau glacée. On acidifie ensuite par un peu d’acide acétique et titre par l’iode décinormal en présence d’empois d’amidon. On répète l’opération sans traitement au formol et obtient le total hyposulfite + sulfite. Par différence on a la teneur en sulfite (Atkinson et Shaner). Un mode particulier de dosage de l’hyposulfite consiste à profiter de sa propriété de ralentir la formation de violet de Lauth par oxydation de paraphénylènediamine par le chlorure ferrique en présence d’un sulfure. On peut ainsi doser chronométriquement I mg d’hyposulfite (66). On peut enfin microdoser le thiosulfate par impulsion de courant selon une technique de Davanathan, Fernando et Peries (Anal. Chim. Acta, 1957, pp. 292-293).
Alun (d’aluminium ou de chrome) : se dose par précipitation à l’ammoniaque, filtration de l’hydroxyde, lavage par une solution chaude de nitrate d’ammonium et calcination à l’état de Al203 ou Cr203.
Les normes ASA spécifient que l’alun ordinaire doit titrer 99,5 % au moins, avec 0,015 d’insoluble, la quantité de fer étant inférieure à 0,04 % et celle des métaux lourds (Pb) inférieure à 0,005 %. Pour l’alun de chrome titrant au moins 98,5 %, à 0,015 d’insoluble, même teneur limite en fer.
L’acidité totale se titre à la soude en présence de vert de bromocrésol ou de bleu de bromothymol. L’acide borique peut être dosé après précipitation de l’alun par l’ammoniaque et de l’hyposulfite et du sulfite par le chlorure mercurique. La solution est filtrée et soumise à l’ébullition pour chasser l’acide acétique. On ajoute un excès d’acide sulfurique, neutralise au vert de bromocrésol et finalement titre par la soude après addition de glycérine.
Acétate de sodium : 50 ml de fixateur sont précipités par le chlorure mercurique ; on s’assure par l’iode et l’amidon de l’absence de sulfite, 15 ml d’acide phosphorique sont ajoutés à la solution qui est distillée. L’acide acétique est titré dans le distillat à la soude en présence de phtaléine
Bromure. : Le fixateur traité par l’ammoniaque pour éliminer l’alun est bouilli, filtré et précipité par le nitrate mercurique ou de plomb. Après nouvelle ébullition suivie de filtration on acidifie par l’acide nitrique, ajoute une quantité connue de nitrate d’argent et on titre l’excès au sulfocyanure d’ammonium en présence d’un sel ferrique.
Sels ammoniacaux : Le fixateur, alcalinisé à la soude, est distillé et les vapeurs recueillies dans une solution d’acide borique. Le borate formé est ensuite titré par HC1 en présence d’héliantine.
Sulfates : Après précipitation de l’alun par l’ammoniaque, et neutralisation, les sulfates, le sulfite et l’hyposulfite sont précipités par un sel de plomb. Connaissant les quantités de sulfite et l’hyposulfite, on trouve celle de sulfate.
Méthode polarographique : Shaner et Sparks ont élaboré une méthode de dosage des aluns, du sulfite et de l’argent, par polarographie (67). L’alun doit être d’abord séparé du fixateur, par précipitation à l’ammoniaque.
Récupération de l’argent des bains d’hyposulfite
Il y a plusieurs méthodes pour récupérer l’argent contenu par les bains de fixage usés (de l’ordre de 5 g/1). Cependant, un fixateur, évaporé au quart, laisse déposer, par cristallisation, de l’hyposulfite presque pur. Tout l’argentithiosulfate reste dans les eaux-mères. Celles-ci peuvent alors passer à la récupération du métal.
1° Précipitation par le zinc. Le zinc déplace l’argent de ses sels : il se forme le sel de zinc correspondant et de l’argent métallique qui précipite lentement. La solution de fixage abandonnée à l’évaporation dans un bac peu profond est ensuite traitée par du zinc en feuille (2 dm2/1 de fixateur). Un dépôt abondant noir se dépose sur le zinc, et du soufre précipite. Le dépôt métallique est détaché à l’aide d’une brosse toutes les 24 h. Au bout d’une dizaine de jours, l’opération est terminée (68). On peut encore neutraliser le vieux bain, le réacidifier avec 2,5 ml de H2SO4 par litre et précipiter l’argent par 200 g de zinc en grenailles (par litre) placé dans un sac de toile.
Le précipité, formé d’argent et de soufre, est recueilli, lavé et séché. On peut le fondre, en présence de carbonate de sodium, ou dissoudre l’argent dans l’acide nitrique.
2° Précipitation par les dérivés de la guanidine (69) : L’argent du fixateur est précipité par l’éthylxanthate de guanidine ou l’anthranilate du même composé. Le bain ainsi traité peut être mélangé à du bain neuf et utilisé à nouveau après addition d’alcali pour ramener le pH à 10. Il faut plusieurs grammes de sel de guanidine par litre de fixateur usé.
La guanidine est une base énergique dérivée de l’urée CO : (NH2)2 dans laquelle O a été remplacé par le groupe NH, soit C(NH) : (NH2)2. Elle se forme par décomposition, à 180°, du sulfocyanate d’ammonium SCN(NH4).
Le chlorhydrate d’hydroxylamine précipite de même l’argent, en solution caustique et à chaud.
3° Précipitation par le sulfure de sodium : C’est la méthode la plus couramment employée. Le bain de fixage est d’abord neutralisé par le carbonate de sodium : l’alun, s’il existe, précipite.
On ajoute ensuite une solution à 20 % de sulfure de sodium Na2S, jusqu’à cessation du précipité. Comme il faut éviter d’ajouter un excès de réactif, on opère par additions successives après essai préliminaire sur la solution surnageant le précipité de sulfure d’argent. Le sulfure de sodium, peut être avantageusement remplacé, selon Koizumi, par du polysulfure d’ammonium en excès (Rt 96 %). Après décantation et filtration, le précipité est séché. Il contient 750 à 800 g d’argent par kilogramme.
Pour extraire le métal (70), on mélange le sulfure d’argent de son poids de nitrate de potassium KNO3. Le mélange, introduit dans un récipient en fonte est chauffé à 200° puis enflammé. La masse,devient incandescente. Le sulfure est réduit en argent et sulfate de potassium et il se dégage du monoxyde d’azote.
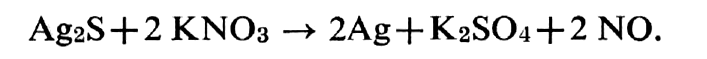
Avant qu’elle refroidisse, la masse est jetée dans l’eau où elle se dissout. Le dépôt est lavé par décantation : il est formé d’argent impur. Par fusion du métal impur avec une petite quantité de borax on obtient de l’argent à 97-99 %. Une autre méthode consiste à griller le sulfure à l’air puis à chauffer le produit obtenu, au rouge blanc, avec du carbonate de sodium et de potassium.
4° Précipitation par l’acide nitrique : Le bain de fixage est concentré au 1/2 ou au 1/3 puis additionné d’acide nitrique à 8 % (en volume). On fait bouillir ce qui précipite l’argent sous forme de sulfure. Le précipité est redissous dans l’acide nitrique bouillant et transformé en chlorure d’argent par l’acide chlorhydrique. On réduit finalement ce dernier par le zinc en milieu chlorhydrique (Miscov, Marinescu et Condurache) (71).
5° Précipitation par l’hydrosulfite de sodium. Le fixateur, amené à pH 8, par neutralisation au carbonate, est additionné d’hydrosulfite : il se produit un dépôt jaunâtre, complet en 12 h. Un fil de cuivre trempé ne doit ni s’argenter (défaut de réactif), ni noircir (excès de réactif).
La précipitation de l’argent d’un bain de fixage, additionné d’un réducteur et d’un alcali, est fortement accélérée par les rayons ultra-violets (Vassiliev) (72).
6° Récupération électrolytique. — La récupération de l’argent des bains fixateurs usés, par électrolyse, permet de remettre ceux-ci en circulation : elle a par contre le désavantage de nécessiter un appareillage important et un contrôle constant des opérations.
Le bain récupéré doit être complété et réacidifié avant de servir à nouveau. Il ne doit pas être utilisé pour le fixage des papiers, qui risqueraient de jaunir.
Un mode opératoire consiste à faire passer un courant électrique dans un bac rempli de la solution à régénérer, préalablement filtrée. Une série de cathodes en acier inox et une série d’anodes en graphite plongent dans le bain agité. L’argent se dépose, bien entendu, sur les cathodes. Tension du courant : 0,3 à 0,5 V. L’agitation est nécessaire car elle évite l’abaissement de la concentration en ions Ag+ au voisinage de la cathode. Spielhagen et Schnôegge (Bild u. Ton, mars 1959, pp. 82-84) préconisent l’emploi d’un bac de 80 1 comportant deux cathodes cylindriques tournant le long d’une anode annulaire extérieure en graphite. Les cathodes sont constituées par des bandes d’argent de 100 mm de largeur distantes de 1,5 mm. Ce dispositif, qui évite la formation de sulfure aux basses densités, permet d’abaisser la teneur en argent du fixateur à 1 g/1, pour un débit continu horaire de 601.
Pour les installations importantes on utilise généralement de fortes densités de courant (73). Pour les installations de faible importance on a de meilleurs résultats en ne travaillant qu’à 1 – 2 mA/dm2. Dans le procédé « Purhypo » élaboré par Doffin (74), la cathode est un gril en acier inox et l’anode une baguette de charbon (Br. Fr. 813.483). L’avantage des faibles densités de courant est qu’elles rendent l’agitation inutile.
Le bain de fixage peut se colorer pendant sa régénération. Cette coloration, qui prend naissance à l’anode, est due à la présence de substances développatrices. Elle est négligeable quand la densité de courant est faible et quand le bain contient du bisulfite (75).
Rasch et Crabtree (76) opèrent à densité de courant moyenne, sans agitation. Avec un bain à l’hyposulfite de sodium acide contenant 8 g de sulfite et 0,5 g d’argent par litre, la densité de courant est 0,01 à 0,02 A/dm2. Avec l’hyposulfite d’ammonium 0,02 à 0,04 A. Le sulfite empêche les réactions en chaîne et la formation de sulfure. La faible quantité de gélatine qui se dépose à la cathode prévient également la sulfuration.
Dans tous les cas, la régénération électrolytique nécessite un contrôle de la concentration du bain, de son pH, de sa température, et s’il y a lieu de son agitation, d’autant plus rigoureux que la densité de courant est plus élevée.
7° Electrolyse sans source de courant extérieure : On peut se dispenser d’une source de courant extérieure si on utilise une électrode en acier inox et une électrode de zinc reliées par un circuit extérieur comportant un rhéostat et un milliampèremètre (77). L’électrode de zinc est enveloppée d’un tissu ou d’une pâte de cellulose. Pour une dimension de 16 X 22 cm immergés, elle peut débiter 0,5 A pendant 48 h, mais il suffit de 12 mA/dm2. Il faut 22 g de Zn pour IO g d’Ag. La majeure partie de l’argent se dépose sur la cathode en inox ; on l’en sépare en imprimant de légères flexions.
8° Récupération par échange d’ions : Les résines contenant des groupes ammonium quaternaire type Dowex 1, absorbent les argentithiosulfates sans les décomposer. Une élution par une solution de chlorure de sodium à 6 % permet de les récupérer. Si la basicité de la résine est trop forte, il y a 50 % de perte par décomposition du complexe (Lindeman et Rabek (78)).
9° Récupération de l’iodure par précipitation par le sulfate thalleux (79) sous forme d’iodure thalleux : Le bromure précipite après l’iodure. Cette opération ne présente pas d’intérêt pratique.
Séchage
Après essorage, mécanique ou pneumatique, l’élément photographique, développé, fixé et lavé, est séché dans un courant d’air filtré. La quantité d’eau retenue par la gélatine est de 0,5 à 2 g/dm2 suivant l’épaisseur de la couche et la nature de la gélatine. En général, 1 000 m de film négatif absorbent 3 000 g d’eau ; les films positifs à couche mince, moitié moins.
Pour les papiers photographiques, il faut compter, en plus, l’eau absorbée par le support, ce qui retarde de beaucoup le séchage.
La température de séchage est comprise entre 21° et 28°. On arrête l’opération quand la gélatine contient encore 15 % d’eau ; autrement elle deviendrait cassante. Dans le séchage sur machine, on redonne au film une certaine humidité.
Pour un degré hygrométrique de 60 à 75 %, le séchage des films de cinéma nécessite 15 à 40 mn pour les films négatifs et 8 à 20 mn pour les films positifs. Le séchage doit, bien entendu, être précédé d’un essorage soit pneumatique (soufflage ou aspiration, soit mécanique avec un tampon de coton). Un film ciné négatif essoré contient encore 375 g d’eau par 100 m et un film positif 150 g/100 m. La vitesse du séchage dépend de l’épaisseur de la couche de gélatine (donc du tannage) et de la vitesse de renouvellement de l’air et de son humidité relative. Un séchage trop rapide peut amener une frisure des bords et des marques de gouttes d’eau.
En cas de nécessité, il est possible de procéder au séchage rapide d’une plaque en plongeant celle-ci dans l’alcool à 80 %, pendant 3 mn, l’égouttant et l’exposant à un courant d’air chaud. Pour éviter les dangers d’incendie, il a été proposé d’ajouter à de l’alcool à 90 %, 2,5 fois son poids d’un fréon à haut point d’ébullition. On évite ainsi, de plus, la formation de taches au séchage.
Un autre expédient consiste à plonger le cliché dans une solution concentrée de carbonate de potassium à 1 000 g/1; après essorage, on essuie et polit la surface gélatinée. Le phototype ainsi traité ne se conserve pas indéfiniment ; à la première occasion, il faut le laver et sécher normalement.
Déformations : L’image photographique séchée peut présenter deux sortes de déformations : les unes sont dues à la distension et contraction du support souple, les autres résultent du travail interne de la gélatine pendant le séchage de l’émulsion vierge et de l’élément développé. Les tensions exercées sont d’autant plus importantes que la couche est plus épaisse.
Un séchage trop rapide, la présence de gouttes d’eau, déterminent des irrégularités très nettes. Le tannage atténue les déformations, car la gélatine se gonfle moins ; par contre s’il y a quand même déformation elle est irréversible, alors qu’avec une couche non durcie on a la possibilité de faire revenir la gélatine à son état normal, par immersion dans l’eau puis dans l’alcool.
Ces distorsions sont gênantes en photogrammétrie. Pour les éviter, dans une certaine mesure, on plonge la plaque vierge dans l’eau, pendant une demi-heure, puis on la sèche après l’avoir lavée à l’alcool. Le séchage doit être lent et dans l’air humide.
Les erreurs dues aux déformations de la gélatine sont de l’ordre de 1 à 4 ,u.
Le retrait dû au support des films cinématographiques est de 0,06 à 0,2 %.
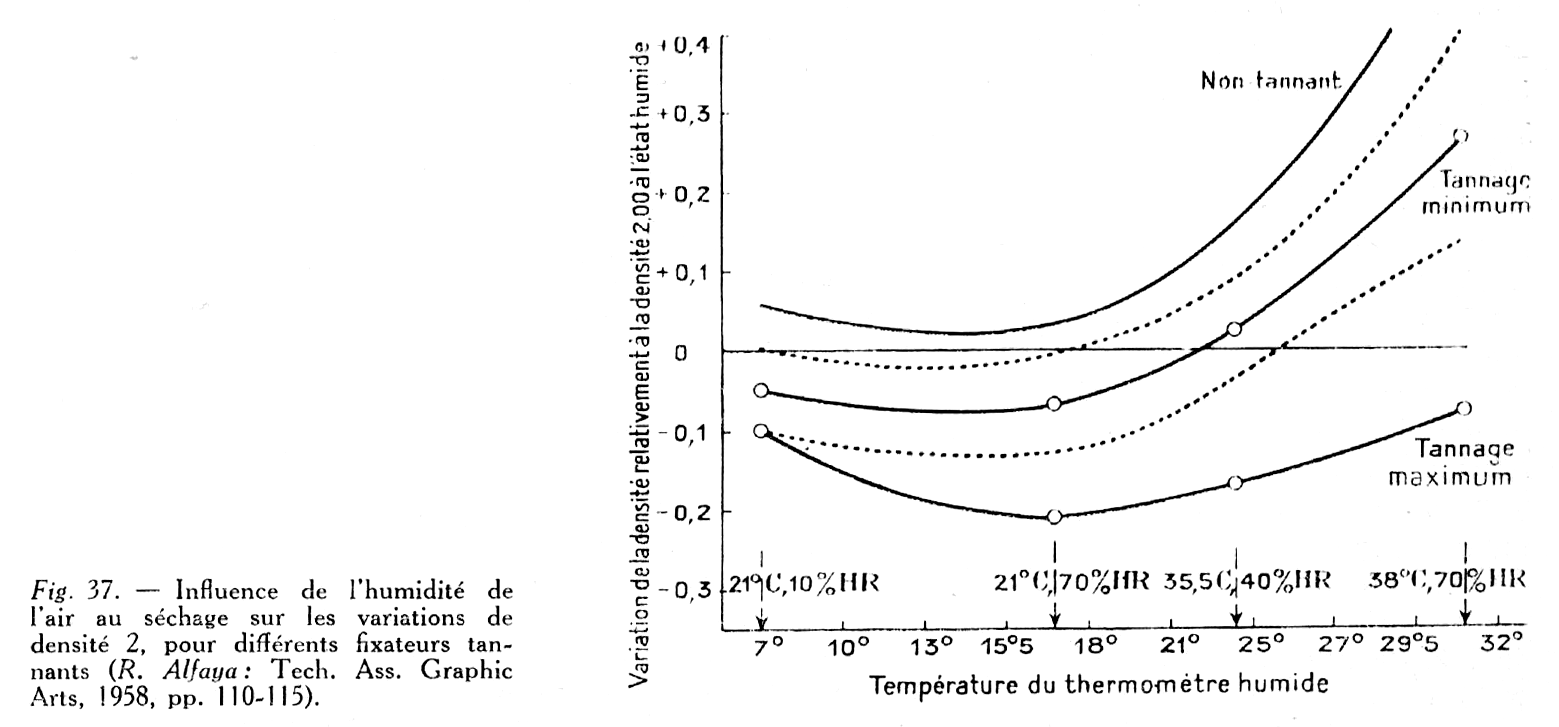
Influence du séchage sur la densité : R. Pinoir a montré (80) que la température et le degré hygrométrique de l’air de séchage influent considérablement sur la densité, par modification de l’état physique de la gélatine. Les résultats dépendent aussi du degré de pouvoir tannant du fixateur (fig. 37).
Une élévation de température et celle du degré hygrométrique augmentent la densité finale, alors que ces deux facteurs agissent en sens contraire, l’un vis-à-vis de l’autre, en ce qui concerne la vitesse de séchage. Par exemple, une densité de 3,08 obtenue à 30° et 75% d’humidité tombe à 2,35 à 22° et 35% d’humidité, soit une différence de 0,73.
Une émulsion gonflée (par exemple juste après étendage et figeage) n’oppose qu’une faible résistance à l’expansion des filaments d’argent développés, mais par séchage la contraction est d’autant plus marquée (Zwicky (81) ). L’effet est maximum sur les gros grains. Il est moins prononcé sur les couches tannées. Il impose de toute façon le contrôle rigoureux du séchage des sensitogrammes et même des films (zones).
L’action du séchage sur la densité s’explique soit par l’hypothèse de la rotation des grains, de Silberstein, soit par l’hypothèse de la compression des grains qui, on le sait, ont une structure spongieuse. La cause de la diminution de pouvoir couvrant au cours du séchage semble être bien due, suivant Blake et Meerkamper (82), à la compression irréversible des grains d’argent, par suite de la formation de liaisons hydrogène dans les molécules de gélatine. Quand un film est mouillé, il y a une rotation des grains tabulaires qui augmente le pouvoir couvrant, mais la compression prédomine dans son effet, sur la rotation, au séchage.
Séchage rapide des films et des papiers, qui a fait de grands progrès ces dernières années, est dû à la nécessité de projeter, immédiatement après la prise de vue, des images de radar et des films de télévision, sans parler de nombreuses autres applications. Le traitement ultra-rapide s’applique surtout aux films spéciaux à couche tannée très mince.
Nous avons déjà vu que le développement et le fixage à température élevée (50 à 60°) permettent le traitement des couches sensibles, en quelques secondes (83). Ainsi Tuttle et Brown (84) ont pu opérer le cycle complet en 4 s (développement 0,8 s, fixage 1,7 s, lavage 0,3 s, séchage 1,2 s). A la température de 60°, avec des liquides ruisselant à raison de 50 ml/s, et en supprimant le fixage ils arrivent même à une durée totale de 0,34 s. Un film ainsi traité peut se conserver, s’il est bobiné serré, pendant une semaine. Après il faut le laver à fond.
Pour la photo sur film radiographique de 70 mm, le traitement total peut, sans inconvénient, durer 1 mn, en accordant 15 s à chaque opération. Température 50°. Le séchage rapide peut se faire au moyen de trois méthodes différentes :
a- Par tambour chauffant : 2 s pour un film se déplaçant à la vitesse de 1,5 rnimn (Ives et Kunz). Inconvénient : mauvaise conductibilité du support cellulosique.
b- Par Jet d’air chaud turbulent à 20 m/s et à une température pouvant aller de 50 à 90°. F.D. Miller (85) a construit à cet effet une chambre soufflante comprenant 256 orifices dirigés sur la face frontale du film et 128 sur la face dorsale et dont le débit total est de 2,5 m3 à la minute. A 20 m/s, il faut pour sécher le film Plus X, jusqu’à 10 % d’humidité, 8 s à 93°, 12 s à 65° et 18 s à 52°. A chaque température correspond une vitesse d’air optimale.
Dans la sécheuse Kodak X-Omat les films transportés par des rouleaux sont séchés entre des jets d’air chaud à 49°. Cela permet de sécher un film radio en 78 s.
Les propriétés physiques d’un film séché de cette manière sont excellentes.
La lenteur du séchage naturel est due à la présence d’une couche d’air saturé stagnante, entre la gélatine et l’atmosphère. On arrive à chasser cette couche isolante, par formation de tourbillons violents, — qu’il ne faut pas confondre avec une simple agitation — à l’aide d’un dispositif analogue à celui de Miller.
L. Katz (86) a imaginé un autre système de séchage turbulent dans lequel le film est tendu sur un tambour de 35 cm de diamètre, chauffé intérieurement à 3 kW pour compenser le refroidissement produit par l’évaporation intense. Un ventilateur de 3 CV projette sur le film en mouvement 0,425 m3 d’air par minute, sous une pression de 0,35 kg/cm2. 5 kg d’eau sont ainsi évaporés par heure, à 48°.
c) Séchage par solvant organique : cette intéressante méthode, due à I. Davies et A.K. Soper (87) permet de sécher rapidement — une minute au plus — un film de surface limitée, par exemple une radiographie.
Le film essoré est plongé dans un mélange à 25 % de diacétone-alcool dans l’éther de pétrole (liquide inflammable), et agité doucement. La diacétone enlève l’eau retenue par la gélatine gonflée, et descend au fond du récipient en une couche non miscible à l’éther de pétrole.
Réticulation
La réticulation est un gaufrage superficiel de la couche gélatinée, produit par un changement brusque de température au passage d’un bain à. un autre, par exemple fixateur tiède après eau froide. Ce phénomène est dû à ce que la gélatine gonflée étant soumise à des tensions parallèles au support, on empêche la couche superficielle de se dilater (88). On peut produire artificiellement la réticulation par immersion dans un acide dilué ou dans une solution concentrée de bromure ou d’iodure de potassium. P.C. Smethurst (89) a utilisé le tannage superficiel avec une solution acétonique d’acide chromique à 5 %, le gonflement dans l’acide phosphorique, ou, mieux, une solution alcoolique de formol gazeux.
Les tensions internes ne se produisent pas si on sèche à une température supérieure au point de fusion de la gélatine, c’est-à-dire, sans faire prendre la couche en gelée.
Glaçage et séchage des épreuves
Les épreuves positives sur papier peuvent acquérir un aspect brillant, par contact, pendant le séchage, avec une surface polie. Celle-ci peut être une plaque de verre, un cylindre de métal chromé ou une tôle chromée à profil convexe. La brillance de la surface gélatinée dépend de celle de la surface glaceuse.
Glaçage à froid : L’épreuve lavée est appliquée humide, sur une glace très propre préalablement frottée d’un agent mouillant ou d’un sulforicinate. Le papier sec doit se détacher de lui-même. Ce procédé qui est lent donne cependant les plus beaux résultats.
Glaçage humide à chaud : Il se fait sur des glaçeuses spécialement construites : plans convexes ou rotatives, chauffées à 75°. Les épreuves sont maintenues en contact parfait à l’aide d’une toile tendue qu’il faut périodiquement enlever et laver. La gélatine doit être bien tannée sinon elle fond et colle sur le métal. Un excès de tannage, dû à une fabrication trop ancienne, ou à un excès d’agent tannant lors de la préparation de l’émulsion, provoque par contre l’apparition de picots sur l’image. La gélatine d’un papier photographique récemment fabriqué (moins de 2 semaines) risque de fondre sur l’appareil. Il est conseillé d’humecter les épreuves lavées avec un produit mouillant, avant de les étaler sur la plaque chauffante, pour favoriser une adhérence uniforme.
Un défaut de barytage du papier peut provoquer des cloques lors du glaçage.
Glaçage à sec et à chaud des épreuves photographiques est beaucoup plus rapide que le glaçage humide. Bien que donnant un brillant moins parfait, il est utilisé pour les travaux industriels comme le tirage des cartes postales.
Il n’est possible que si la gélatine contient 10 à 15 % d’humidité, et si la température est supérieure à 80°. Les meilleurs résultats sont obtenus entre 110 et 120°.
La température critique optimum dépend de la nature de la gélatine et de son degré d’humidité. Trop humide la gélatine colle ; trop sèche elle n’acquiert aucun brillant. Si le cylindre est trop chaud, elle jaunit. De nombreux autres facteurs inconnus interviennent sans doute, car le glaçage à sec est généralement une source d’ennuis « inexplicables ». Les dépôts calcaires peuvent eux aussi atténuer la brillance. Par ailleurs, un cylindre trop propre fait coller les épreuves, contrairement à ce que l’on croit généralement. Toute application d’agents mouillants, savons ou solvants organiques sur la face gélatinée avant glaçage à une action néfaste.
Séchage sur glaceuse : Une épreuve photographique placée sur une glaçeuse, gélatine contre la toile, sèche sans devenir brillante. On peut ainsi traiter les papiers artistiques à surface mate et les papiers pour documents. Une gélatine trop tendre ou récemment coulée colle sur la toile, ce qui oblige à arracher le papier. Si la pâte même du papier n’a pas une structure isotrope, l’épreuve sèche se roule ou se gaufre d’une façon irrégulière.
Examen analytique des dépôts formés sur les images
R.W. Henn (90) a établi un mode opératoire pour l’identification de la nature des taches et dépôts, accidentellement présents sur les images photographiques. L’essai est fait par touche sur un morceau de rebut. On dépose deux gouttes de solvant sur la tâche. Après dissolution, une goutte du réactif est transférée sur une plaque de verre ou de porcelaine, pour être soumise au réactif d’identification.
- Calcium : Solvant : acide acétique à 40 %. Neutraliser par AmOH. Précipiter par oxalate de sodium à 4 %. Ou encore dissoudre la tache dans HCI chaud. Verser 2 gouttes de AmOH à 10 % puis 1 goutte d’alizarine S à 0,1 %. Il se produit, avec Ca, une coloration bleue et des cristaux visibles au microscope.
- Aluminium : Solvant : acide acétique à 40 % ou (soude+ sulfate de sodium) à 10 %. Ajouter 1 goutte de solution alcoolique de Morin (puis 1 goutte d’acide acétique si le solvant est de la soude) : coloration brun à. jaune-verdâtre. La laque obtenue est fluorescente en lumière UV ou à la lumière du soleil filtrée par un filtre Wratten 35.
- Chrome : ,Solvant : HCI dilué, ajouter 1 goutte d’eau de brome. L’ion chromique formé est neutralisé par la soude, l’excès de brome par du phénol et on ajoute de la diphénylcarbazide en solution alcoolique à 1 % ainsi que H2SO4 2 N : il se forme une coloration bleu violacé.
- Fer : Taches jaunes, solubles dans l’oxalate de potassium à 5 % additionné de 5 % de HCI. Taches bleues, décolorées par la soude.
La solution chlorhydrique précipite en bleu par le ferrocyanure de potassium. - Argent : Taches jaunes ou brunes souvent dichroïques, solubles dans le ferricyanure+ hyposulfite. La solution nitrique des taches précipite par l’acide chlorhydrique. Egalement, réaction à la diméthyl-benzalrhodanine.
Auteur Pierre Glafkidès
Physique et chimie photographique
56- Hickman et Spencer : Sc. Ind. Phot., (1) t. 3R, p. 109 et t. 5R, p. 5.
57- Jelley et Clark : Sc. Ind. Phot., t. 1 (2), p. 57 et 253.
58- Crabtree et Ross : Sc. Ind. Phot., t. 1 (2), p. 215.
59- Przybylowicz, Zuehlke et Ballard : Phot. Sci. Eng., octobre 1958, pp. 148-153 ; Sc. Ind. Phot., février 1959, pp. 63-65.
60- E. Cary et H.H. Wheeler (U.S. Dept. of Agriculture) : Amer. Phot., février 1942, pp. 16-18
61- Cette méthode préconisée par Gibson et Weber (Se. Ind. Phot., t. 10 (2), p. 120) a été adoptée par la norme ASA PH. 4-8-1953. (Sc. Ind. Phot., novembre 1954, p. 441).
62- J.l. Crabtree, G.T. Eaton et L.E. Muehler Il. Franklin Inst., avril 1943, pp. 351-360.
63- R.W. Henn et J.I. Crabtree : Phot. Sc. Techn., août 1954, pp. 83-84 et Se. Ind. Phot., novembre 1954, p. 441.
64- R.W. Henn et J.1. Crabtree : Phot. Sc. Techn., août 1955, t. 2, pp. 111-113 et Sc. Ind. Phot., novembre 1955, t. 26, pp. 447-448..
65- Br. Fr. 825.306 (1936).
66- J.B. Risk et D.H. Strickland : Analyt. Chem., mars 1957, pp. 434-437.
67- V.C. Shaner et Miss M.R. Sparks : JI. of Mot. Pict. Eng., juillet 1945, pp. 20-32 et Sc. Ind. Phot.,1945, nos 9-10.
68- Sedlaczek a préconisé l’emploi de poudres métalliques en présence de dérivés de l’acide sulfocarbonique (Br. All. 713.056).
69- Br. Amér. 2.221.163.
70- Stratter : Chem. e Industrie (Milan) 1941, t. 23, n° 6, p. 213.
71- Miscov et Coll.: Farmacia, 1959, pp. 167-169 (en roumain).
72- Vassiliev : J. Prikl. Khim., mars 1954, pp. 258-264.
73- Hickman : Sc. Ind. Phot., t. 2 (2), pp. 462-470.
74- C.J. Sharpe : Sc. Ind. Phot., mai 1952, p. 185. — Voir aussi H.G. Doffin : Sc. Ind. Phot., t. 10 (2), p. 182.
75- C.J. Sharpe : Brit. JI. Phot., mars 1954, p. 140.
76- A. Rasch et J.I. Crabtree : Pitot. Sei. Techn., février 1955, t. 2, pp. 15-33 ; Sc. et Ind. Phot., avril 1955, t. 26, pp. 137-142.
77- K. Heiberg : Acta Radio!, septembre 1950, pp. 215-224, et Sc. Ind. Phot., avril 1951, p. 133.
78- J. Lindeman et T. Rabek : Chem. Techn., 1959, pp. 147-149.
79- K. Kieser : Phot. Ind., t. 38, février 1940, p. 96. — L.G. Welliver : Sc. et Ind. Phot., avril 1956, t. 27, pp. 144-145.
80- R. Pinoir : Sc. Ind. Phot., novembre 1943, p. 241.
81- H. Zwicky : Zeits. wiss. Phot., décembre 1955, pp. 415-424.
82- K. Blake et B. Meerkamper : JI. Phot. Sci., janvier 1961, pp. 14-25.
83- J.I. Crabtree : JI. Phot. Soc. Amer., février 1949, pp. 130-136, et Sc. Ind. Phot., 1949, p. 216. — lves et Kunz :.11. Sc. Instrum., 1951, pp. 318-319, et Sc. Ind. Phot., janvier 1952, p. 21.
84- C.M. Tuttle et F.M. Brown : Jl. of Mot. Pict. Eng., février, pp. 144-160, et Sc. Ind. Phot., juin 1950, p. 223 ; Phot. Eng., juin 1952, pp. 65-77.
85- F.D. Miller : JI. of Mot. Pict. Eng., février 1953, pp. 85-104, et Sc. Ind. Phot., juillet 1953, p. 288.
86- L. Katz : J’1. of Mot. Pict. Eng., mars 1951, pp. 264-279, et Sc. Ind. Phot., août 1951, p. 309.
87- I. Davies et A.K. Soper : Brit. J1. Phot., mai 1950, p. 268, et Sc. Ind. Phot., août 1950, p. 295.
88- Voir étude par Sheppard et Elliot : Ind. Eng. Chem., 1918, p. 727.
89- P.C. Smethurst : Brit. JI. Phot., août 1949, pp. 371-374, et Procédé, février 1950, pp. 1-5.
90- R.W. Henn : J!. Phot. Soc. Amer., cité par Sc. Ind. Phot., août 1951, p. 299.